
|
||||||
|
|
|
|
Download | |||
Diamond Developer BlogExpanding to and reducing from a molecule clusterKlaus Brandenburg -- 8 May 2013, 9:00 UTC This article shows how to expand from a set of atoms or a molecule via neighbouring molecules to a cluster and to reduce that cluster back to a smaller ensemble of molecules again. You can do this with or without H-bond and/or contact definitions -- starting from a selected atom or a selected molecule or from the whole current ensemble in the structure picture. We use the compound COD:1500005 (hexaammine cobalt(III) hydrogenarseniate tetrahydrate; Ritu Bala, Raj Pal Sharma, Rajni Sharma, Juan M. Salas, Miguel Quiroos, William T.A. Harrison; J. Mol. Struct. 828 (2007), 174-180) to demonstrate that. We start with the command "Create molecules directly" (via accelerator key Ctrl+M or clicking the corresponding symbol in the Picture toolbar) and get ten molecules created from the atomic parameter list. 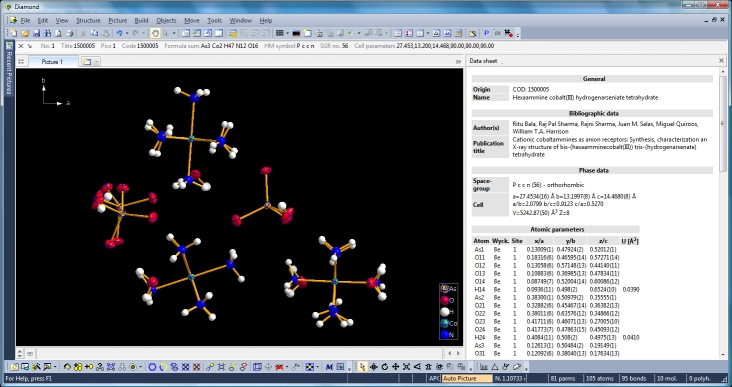
Switching from data sheet in the right half of the Diamond application window to the table of created molecules ("View" menu, "Table" sub-menu) we get a list with molecule titles and formula sums and how far the molecules are complete or still fragmentated. Since we will put "Molecule 4" into the focus of our interest, the corresponding row is highlighted and the properties shown in the "Properties" pane below. 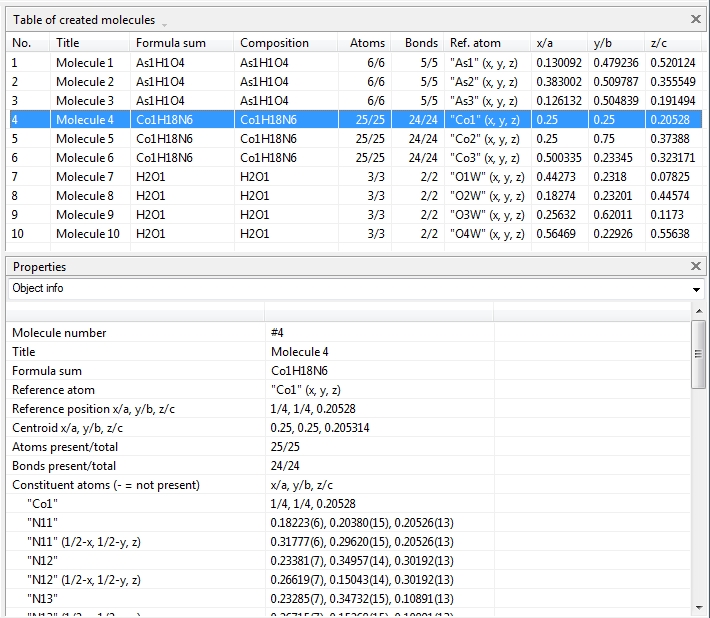
Clicking with the right mouse button on the "Molecule 4" row in the table of created molecules and choosing command "Select Reference Atom" marks the atom "Co1" (at x,y,z) in the lower left of the structure picture. Running command "Edit"/"Select Molecule" (or accelerator Ctrl+M), followed by "Edit"/"Invert Selection" (or Ctrl+I), followed by "Edit"/"Delete" (or "Del" key) leaves the "Molecule 4" alone in the picture, which will serve as starting point for our expansions and reductions. 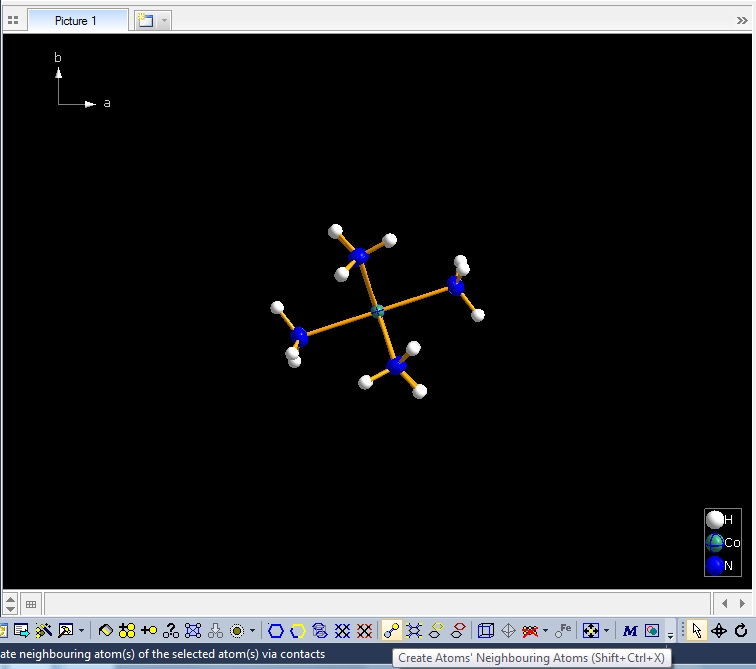
Clicking the button (highlighted in the bottom center of the screen shot above) "Create Atoms' Neighbouring Atoms" or simply pressing Shift+Ctrl+X (for "eXpand") three times leads to the following sequence: 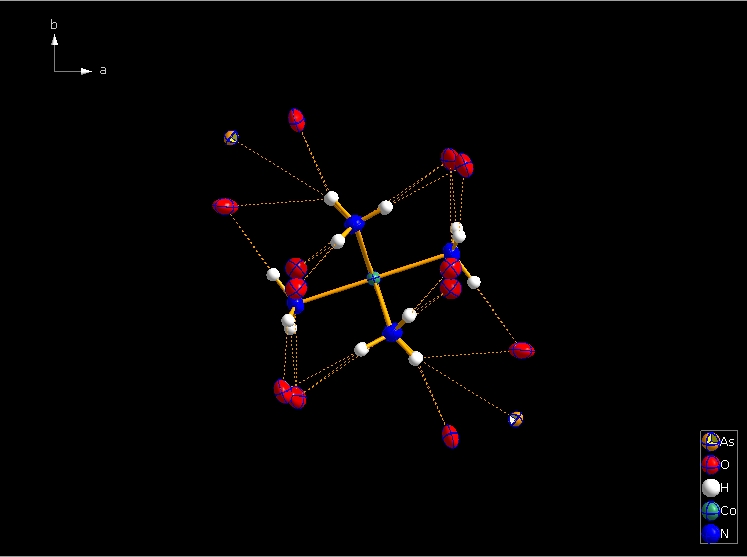 Contacts from the atoms of the molecule to neighbouring atoms (depicted as dotted lines). 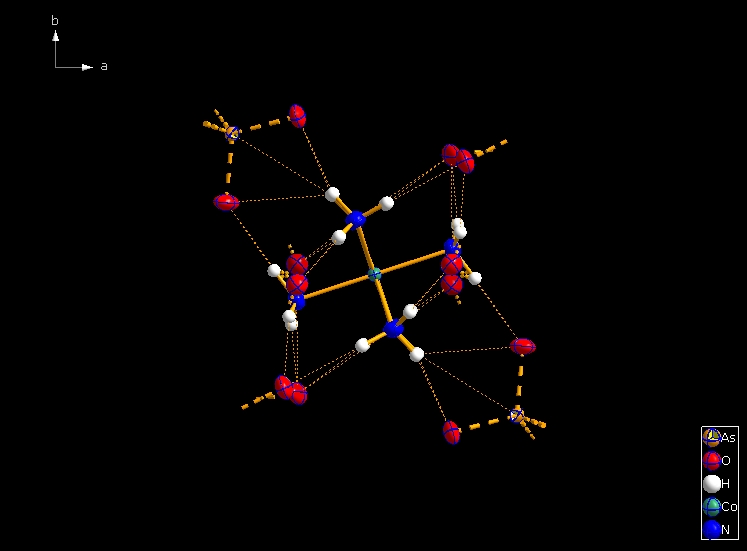 Next "Expand" command creates broken-off bonds to the next bonded atoms of the neighbouring molecules each. 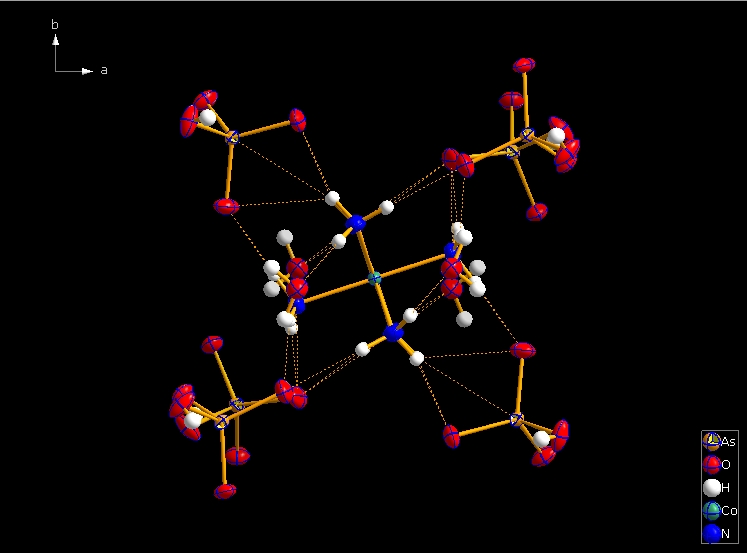 Running "Expand" again completes these fragmentated molecules. If you want to, you may start another round finding neighbouring molecules. The example above applies to the whole molecule "Molecule 4", but you could have marked a special atom of the molecule, for instance one of the N atoms rather than the "Co1" in the center, to show selectively the neighbourhood of that N atom. Finding and creating neighbouring molecules goes quicker with the "Create Neighbouring Molecules" symbol in the toolbar (or Shift+Ctrl+Z). Two runs of the command lead to the following screen shot: 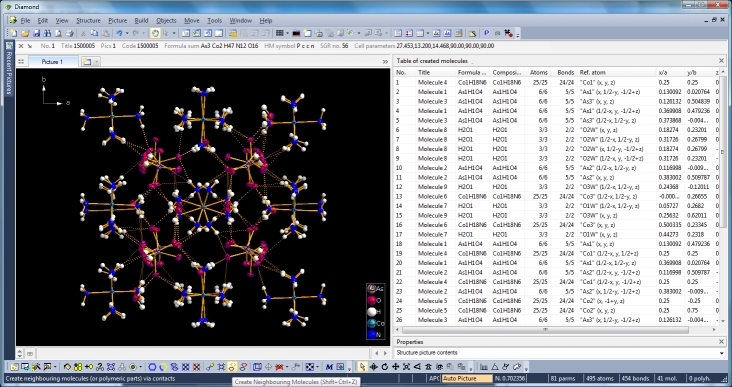
You can dismantle a molecule cluster step-by-step using the "Reduce" command (either from the "Build" menu, giving you several options, such as where to start from -- a selected atom/molecule or from the center of the picture) or simply clicking the button highlighted at the bottom of the next screen shot: 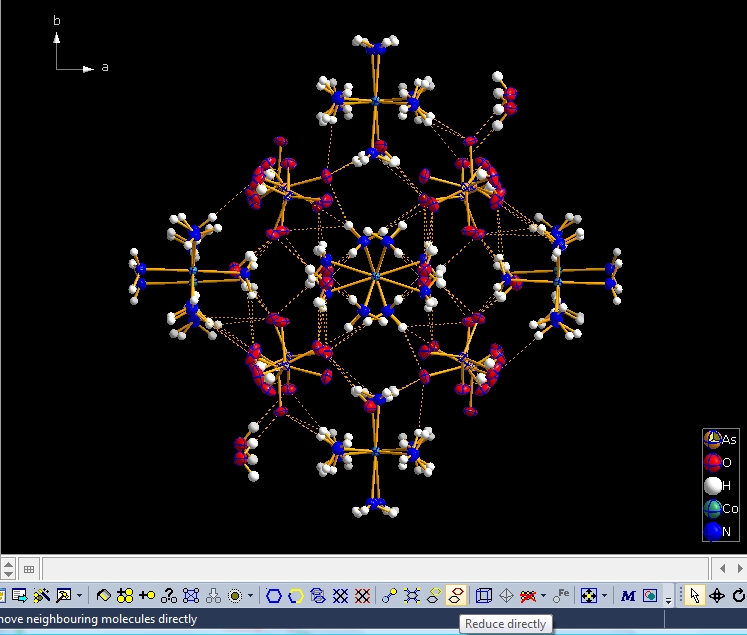
And repeating "Reduce" several times leads you finally back to a picture with the one and only "Molecule 4". The contact definitions in the example above come from "_geom_hbond_xxx" definitions in the 1500005.cif file. But there are several possibilities to change, add, or remove definitions for non-bonding contacts in the "Connectivity" dialog ("Build" menu). |
|
| Page last modified May 15, 2018.
Copyright © 1997-2018 Crystal Impact GbR. All rights reserved. Imprint / Contact Privacy policy Webmaster Google+ Twitter |