|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Building up structural partsConnectivity, part 1: Evaluation of Bonding SpheresUse the Connectivity dialog to define which atom groups are to be connected
This article in brief:
Previous article: Filter About ConnectivitySince the definition of the bonding spheres in the bond group list is essential for all building functions that generate bonds, Diamond offers a separate and sophisticated way to handle these bonding spheres. (Note: There is an additional criterion for bonding spheres defined on atom site level, called "atomic environments", which is subject of the following article.) For each atom group pair (that means for each bond group) a bonding sphere is defined and a flag whether this bonding sphere is active or not, i.e. whether bonds are allowed for that atom group pair or not. To edit the connectivity list, choose the Connectivity command from the Build menu.
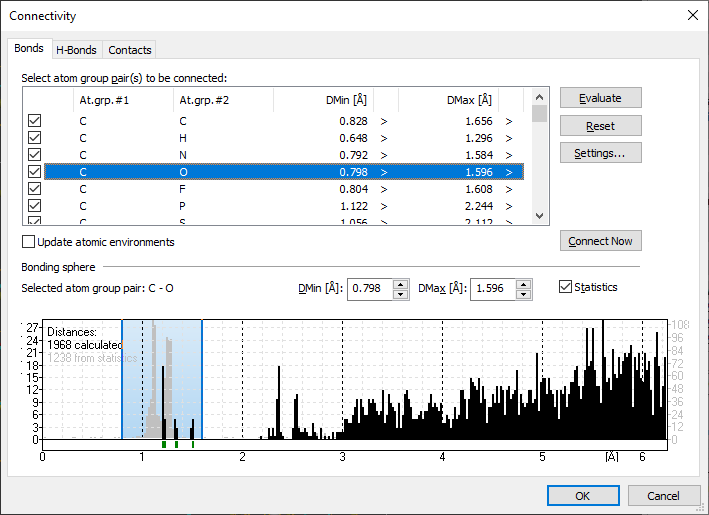
In the Connectivity dialog, you will find a list of all atom group combinations (bond groups).
Those atom type combinations which have been defined as bonded have a checkmark set. The lower half shows the Bonding sphere of the currently marked bond group (atom group pair).
Additionally you will find the lower and upper value for the actually selected bonding sphere as well as the distribution of interatomic distances in the current structure. Default initialization of connectivity list
Whenever a structure has been imported or the atomic parameter list has been edited, bond groups will be initialized or reset, rsp. The bonding sphere of a bond group is calculated like this:
If a mean bond value is defined for the element combination of the bond group (ignoring oxidation numbers) in the factory default settings of Diamond or in the Diamond section of the Windows Registry (if you have made changes to the default value), this value is taken as mean value of the bonding sphere. Otherwise, the effective radii of the two atom groups are added.
The result is multiplied with the lower sphere boundary factor to get the lower boundary value of the bonding sphere. The upper sphere boundary factor is used for the upper boundary value. Diamond works with default boundary factors of 0.6 and 1.2, rsp.
To edit the boundary factors, push the Settings button in the Connectivity dialog, and edit the values in the Settings for connectivity dialog.
All bond groups are selected, unless the oxidation numbers have the same sign.
Enabling or disabling bonding for an atom group combinationIn the list Select atom pair(s) to be connected, click on the checkmark of the atom group pair you want to enable or disable, rsp.
Changing the bonding sphere for an atom group pair
If the atom group pair has been selected in the list Select atom pair(s) to be connected, edit the lower and/or upper value in DMin and DMax rsp. or shift the blue lines in the histogram.
To shift one of the blue lines, move the mouse cursor to the line. (The mouse cursor changes its symbol.) Press the left mouse button and move the line to the desired position. When you have reached the final position, release the left mouse button. The new lower or upper value will be reflected in the DMin or DMax field as well as in the list Select atom pair(s) to be connected.
Please note that the calculation of the interatomic distances to be displayed in the histogram may take a while, especially for structures with some thousands of atoms in the parameter list (e.g. proteins).
Evaluating all bonding spheres
Click the Evaluate button. This will execute the following steps:
(1) All bonding spheres are reset to the initial values (see above: Default initializiation of connectivity list).
(2) For each bonding sphere, interatomic distances are calculated. A bond group will then be selected (checkmark set), if at least one distance is found within the bonding sphere range.
Resetting all bonding spheres
Click the Reset button. This will re-initialize the bonding spheres. (See above: Default initializiation of connectivity list.)
More features of the Connectivity dialog, Bonds page
Consideration of bond parameters
Connectivity dialog has variable size
Atom group pair list indicates changes
Distances from bond parameters as ticks
Previous article: Filter
Reference for COD:7103386: |
|
Page last modified July 7, 2022. Copyright © 2022 Crystal Impact GbR. All rights reserved. Contact Webmaster |