Packing Dialog
There are several options
to create a packing diagram (cell range, sphere, slab, or slice of molecules) and how far to include
molecules
This article in brief:
- Comprehensive two-pages dialog "Build/Packing...".
- Page "Choose Atoms" to choose the molecules to be considered, how to treat polymers,
and if and how to include, exclude or cut molecules at the packing boundaries.
- Page "Define Range" to define the boundaries of the parallel epiped. You may also
define a sphere instead of a parallel epiped.
- Option to fill a slab or slice parallel to an hkl plane (compare command "Build/Fill/Slab"),
with the cell range as delimiters.
- Toolbar button or hotkey Ctrl+Shift+G considers unit cell as range, independent
from the latest dialog settings and includes molecules when centroid inside.
Previous article: Adding and finding symmetry-equivalent molecules
Next article: Inserting (dummy) atoms and bonds
Comprehensive Packing Dialog
The command Packing... in the Build menu opens a comprehensive dialog to
create a packing (diagram), that means to define and fill the contents of a cell
range with molecules (and/or polymers and/or single atoms). The first page "Choose
Atoms" defines, how to treat molecules that cross the packing range's boundaries
and how to deal with polymers and non-bonded ("vagabonding") atoms. The second page
"Define Range" offers a variety of cell ranges (unit cell, super cells or a parallelepiped
in general, a sphere, or a slab or slice parallel to an hkl plane.
"Choose Atoms" page
The page "Choose Atoms" chooses the molecules to be considered, how to treat polymers,
and if and how to include, exclude or cut molecules at the packing boundaries.
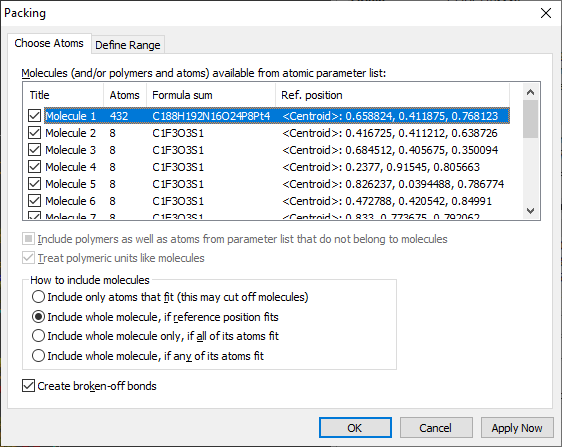
Molecular units etc.
The list under "Molecules (and/or polymers and atoms) available from atomic parameter
list" lists all molecular units that have been generated from the atomic parameter
list, either by current connectivity settings or by current atomic environment definitions.
This can be checked and changed under "Build"/"Molecules"/"Define..." for all molecule
generating and molecular fragment completing functions. The list also contains vagabonding
(non-bonded) atoms as well as polymers, unless you suppress them by clearing the
checkmark at "Include polymers [...]".
How to include molecules
This is the main option on this page, since it roughly defines how many molecules
will appear in your packing diagram. The default and best choice is:
- Include whole molecule, if reference position fits. With this
option, you usually get the number of molecules corresponding with the number of
formula units. And usually the result is a picture, where some molecules have their
feet outside the cell, whereas on the other side, some atoms of the cell are missing,
which means there are typically not all atoms in the cell that a command like "Fill
unit cell" or "Fill cell range" would generate.
- The option "Include whole molecule only, if all of its atoms fit"
assures that no part of the molecule lies outside the packing range. That results
usually in significantly less atoms/molecules than in the first mentioned option.
- On the other side, the option "Include whole molecule, if any of its atoms
fit" usually creates the most atoms/molecules, because one atom is
enough to get the whole molecule considered for the packing diagram. This is roughly
the same as "Fill unit cell" or "Fill cell range" plus a subsequent call of "Complete
fragments" command.
- The option "Include only atoms that fit" is somewhat different.
And actually it is more like "fill cell range" plus "connect atoms" -- plus a "create
broken-off bonds" command call, if the checkmark at "Create broken-off bonds" is
set. The advantage here is that you can choose molecules when you have multiple
molecular units in your structure, since the normal "Fill cell range" functions
do not consider molecular units.
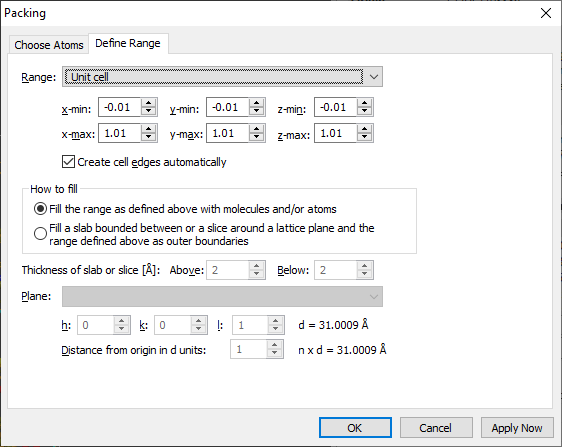
"Define Range" page
The page "Define Range" defines the boundaries of the parallel epiped. You may also
define a sphere instead of a parallel epiped.
Parallelepiped or sphere
Define a parallel epiped with boundaries in crystal coordinates x-min through z-max
or choose the unit cell or one of the pre-defined supercells. Default setting is
a unit cell with boundaries like under "Build"/"Fill"/"Unit Cell". (The offsets
-/+0.01 are to catch the atoms lying on faces and edges or in the corners of the
parallepiped.) Or choose a sphere rather than a parallelepiped and define x/a, y/b,
z/c of the center and minimum and maximum sphere boundaries in Angstrom.
Slab or slice
Instead of parallelepiped or sphere you can fill a slice of thickness Above
+ Below parallel to a plane given by Miller indices h,k,l and distance
d from origin. In that case, the range boundaries are used to delimit the (actually
infinite) slice. For a slab, define a Below with at least n x d Angstrom.
The slab definition is similar to the one in "Build"/"Fill"/"Slab...".
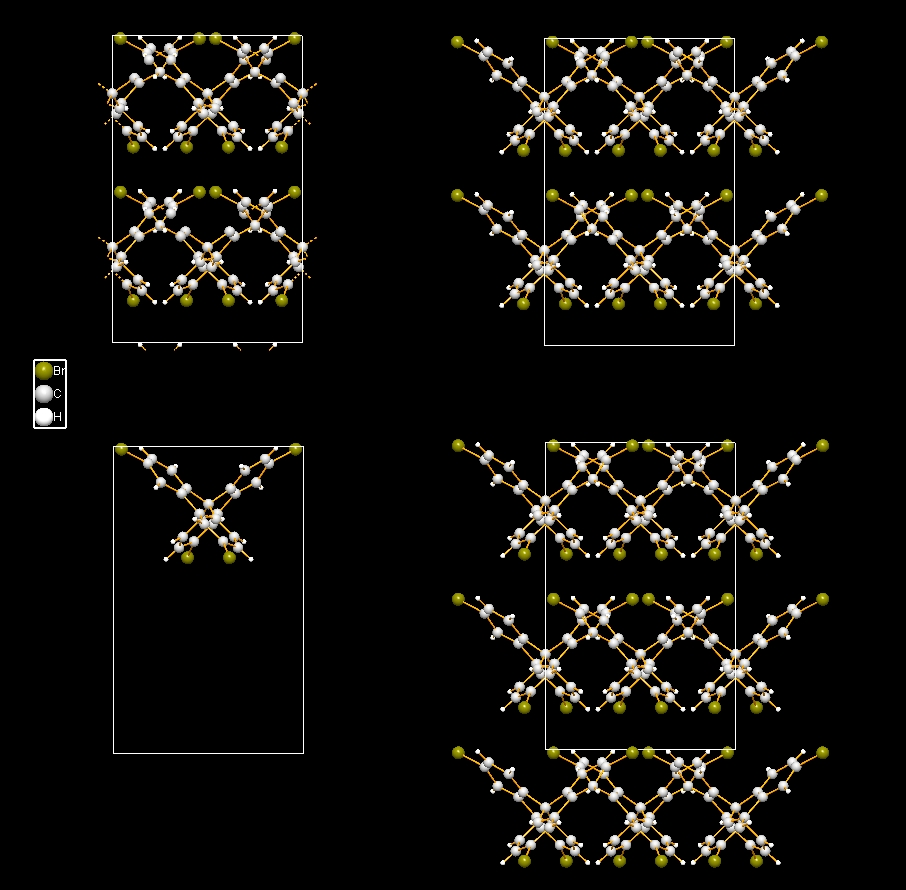
Sample #1
COD:1501635, which has 1 molecular unit only. The following image is a composition
of four structure pictures of unit cell packing from sample "COD-1501635-unit-cell-packings.diamdoc".
- Upper left: Include only atoms that fit (13 incomplete and just one complete molecule).
- Upper right: Include whole molecule, if reference position fits (nine complete
molecules).
- Lower left: Include molecule only, if all atoms fit (just one molecule fits completely
in the unit cell).
- Lower right: Include whole molecule, if any of its atoms fits (14 molecules, all
complete).
To create a packing picture each, first delete all contents ("Build"/"Destroy"/"All"
or Shift+Ctrl+D), then run the "Packing" dialog, choose the right molecule including
option (and care for the right settings for cell range), and close the dialog with
OK.
Sample #2
COD:1500005, Hexaammine cobalt(III) hydrogenarseniate tetrahydrate, which has 10
molecular units:
- "Molecule 1" through "Molecule 3": HAsO4 (hydrogenarsenate).
- "Molecule 4" through "Molecule 6": Co(NH3)6 (hexaammine cobalt-III).
- "Molecule 7" through "Molecule 10": H2O.
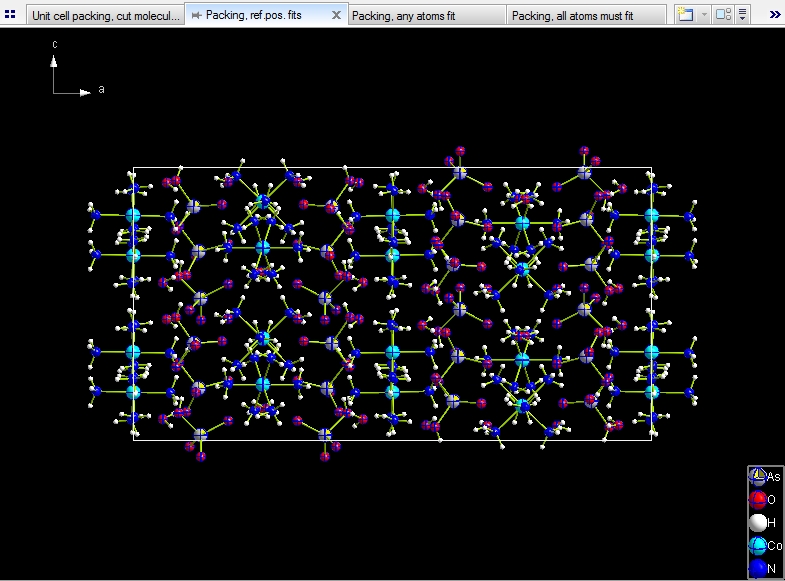
The following pictures show unit cell packings each, viewed along b-axis, but with
four different choices how to include molecules:
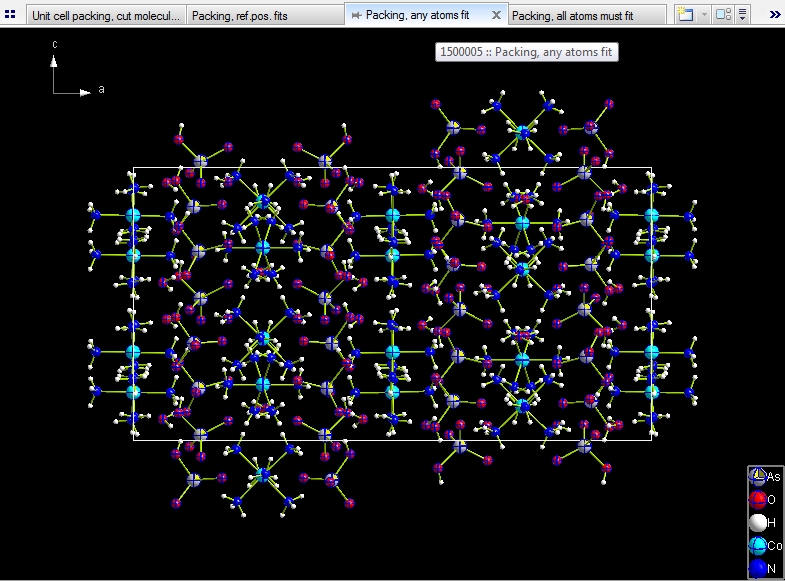
Include only atoms that fit: This results in a total of 102 molecules,
64 of them are complete, because completely within the range boundaries, the residual
38 molecules crossing the boundaries are incomplete (fragmentated).
Note: The screenshots (made with Diamond version 4) use tabs. To activate tabs, use command "View"/"Pictures Viewing"/"Show Tab Bar".
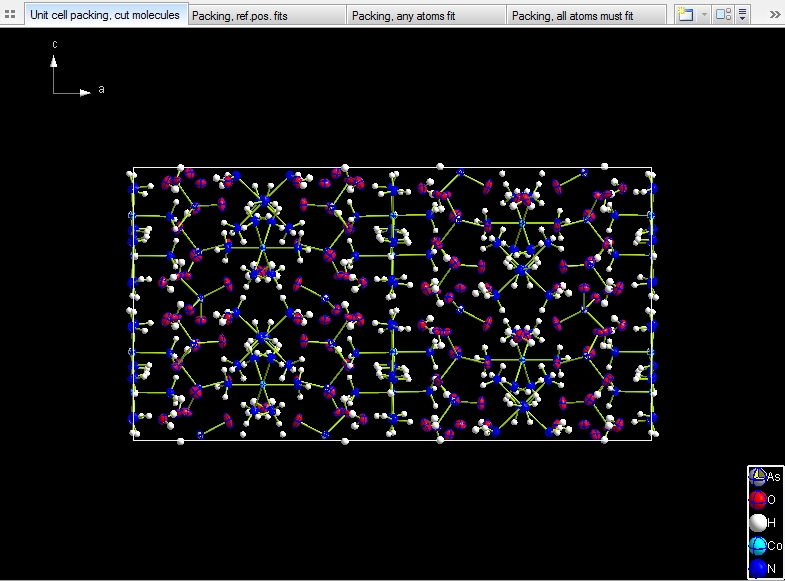
Include whole molecule, if reference position fits. This gives
you the 8 * 10 = 80 (complete) molecules, corresponding with the number of formula
units (Z) of 8 and 10 molecular units.
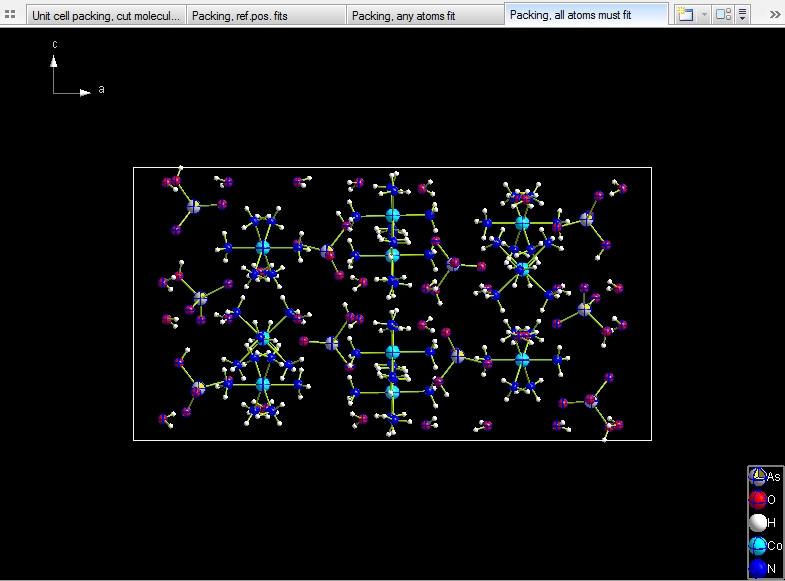
Include whole molecule only, if all of its atoms fit. There are
just 52 molecules.
Include whole molecule, if any of its atoms fit. Like in the first
choice we have a total of 102 molecules, but here they are all complete.
Selectively creating molecules in packing
In many (or most) cases, the atomic parameter list contains more
than just one molecular unit only, and there are often solvent molecules or so.
Here we will show how to select (and unselect unwanted) molecular units, so they
do not appear in the packing diagram.
Open the sample file "COD:7103386.diamdoc", which comes up with a unit cell packing
(which was created using the option to include molecules whose centroid fits), containing
32 molecules -- there are 16 molecular units and Z (number of formula units per
cell) is 2.
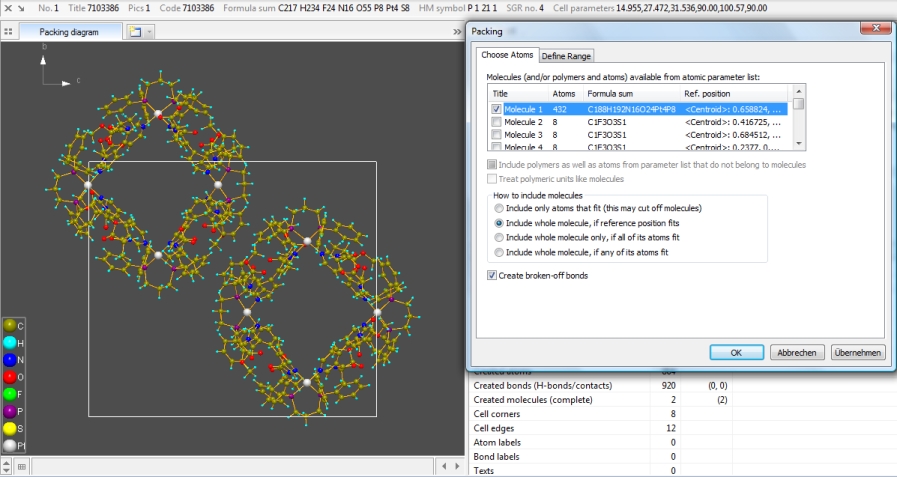
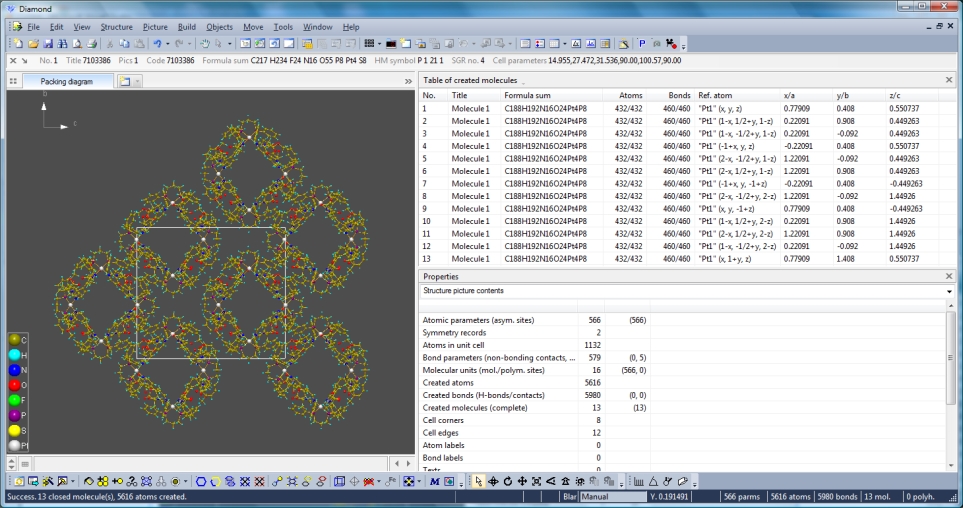
Now we want to drop all molecules but "Molecule 1" having "Pt1" as reference atom
and consisting of 432 atoms (formula sum C188H192N16O24Pt4P8) and 460 bonds. (Cf.
"View" -> "Table" -> "Table of Created Molecules".) That means we will clear
the 15 checkmarks at molecules "Molecule 2" through "Molecule 16" (CF3SO3 and C3H6O).
Ensure that option "Include whole molecule, if reference position fits" is activated
and the range (page "Define Range") is set to "Unit cell". As result we get two symmetry-equivalent molecules of "Molecule 1", one at
reference atom position (x, y, z), the second at (1-x, 1/2+y, 1-z):
Destroying the picture and creating the same unit cell packing for "Molecule 1"
but now with option "Include whole molecule, if any of its atoms fit" results in
the following picture with a total of 13 molecules. The table of created molecules
gives an overview -- at least of the reference positions, the rest is nearly the
same.
Accelerators to create packings
There is a toolbar button and a hotkey Ctrl+Shift+G to create a
packing without involving the "Packing" dialog. This accelerator command considers
unit cell as range, independent from the latest dialog settings, and includes molecules
when centroid inside. (These are also the default options for the "Packing" dialog.)
Previous article: Adding and finding symmetry-equivalent molecules
Next article: Inserting (dummy) atoms and bonds
References:
COD:1501635: Ulrich Darbost, Janie Cabana, Eric Demers, Thierry
Maris, James D. Wuest; Molecular Tectonics. "Use of Br...aryl Supramolecular Interactions
for the construction of Organized Networks from 9,9'-spirobifluorene in the Crystalline
State"; CheM, 1 (2011), 52-12369.
COD:1500005: Hexaammine cobalt(III) hydrogenarseniate tetrahydrate.
Ritu Bala, Raj Pal Sharma, Rajni Sharma, Juan M. Salas, Miguel Quiroos, William
T.A. Harrison; "Cationic cobaltammines as anion receptors: Synthesis, characterization
an X-ray structure of bis-(hexaamminecobalt(III)) tris-(hydrogenarsenate) tetrahydrate";
Journal of Molecular Structure, 828, 174-180 (2007).
COD:7103386: Rang, Alexander; Nieger, Martin; Engeser, Marianne;
Lützen, Arne; Schalley, Christoph A; "Self-assembling squares with amino acid-decorated
bipyridines: heterochiral self-sorting of dynamically interconverting diastereomers.";
Chemical communications (Cambridge, England), *, 4789-4791 (2008).
|
