Endeavour Features: Structure Solution Wizard
This page introduces Endeavour's so-called 'Structure Solution Wizard' for the
easy preparation of the structure solution process.
Endeavour Features Overview...
Next: Space-group Determination...
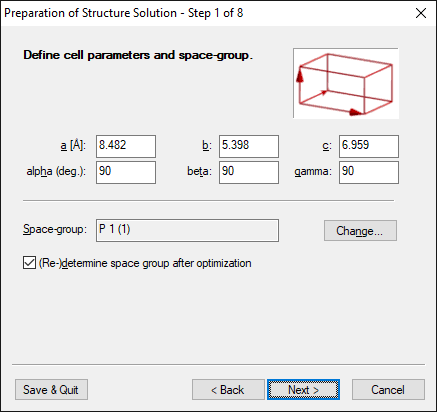 |
Step 1 of 8:
Define cell parameters and space-group
Cell parameters must be known, i.e. the powder data must be indexed, whereas
the knowledge of the correct space-group is not essential. The calculation can
always be performed in P1 and the correct space-group can be determined
afterwards. However, if the space-group is known, the calculation speed is much
higher. |
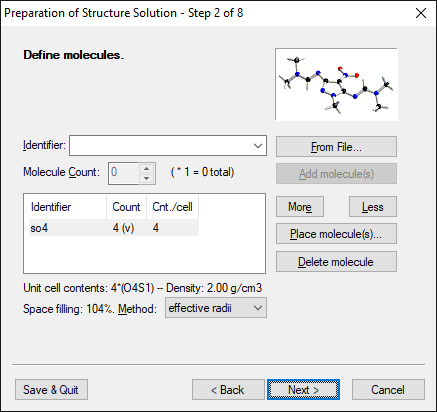 |
Step 2 of 8:
Define molecules (optional)
Endeavour supports the usage of molecules with flexible torsion angles as
well as rigid-body fragments if they do not lie on special positions of the
spacegroup. Molecules can be fixed at certain positions and may rotate along a
reference atom.
|
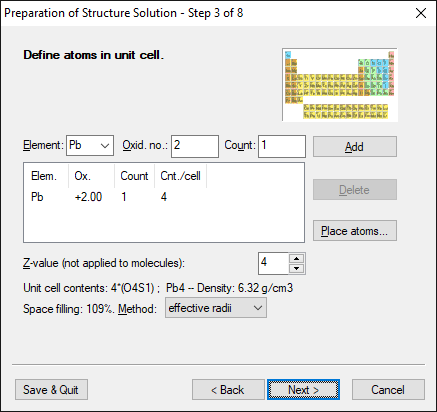 |
Step 3 of 8:
Define atoms in unit cell
Enter the unit cell contents, i.e. the formula sum and the number of formula
units per unit cell for single atoms, i.e. atoms which do not belong to
molecules.
|
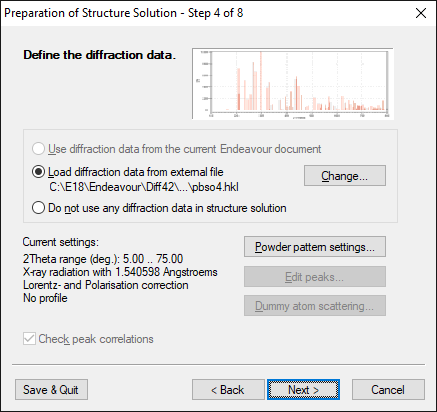 |
Step 4 of 8:
Define the diffraction data
The next wizard page is dedicated to the input of the diffraction data. You can
open the prepared file containing the diffraction data as a list of 2 theta-
and corresponding intensity values, or, as in this case, a list of |F|(hkl)
data.
|
 |
Step 5 of 8:
Choose between automatic and manual structure solution settings
Since version 1.6 all structure solution calculation parameters can be adjusted
automatically after cell parameters, space group, molecules, single atoms, as well
as diffraction data have been entered. This is the default and recommended setting,
and you are led to the final page of the wizard. (In this example we continue with
the next page.)
|
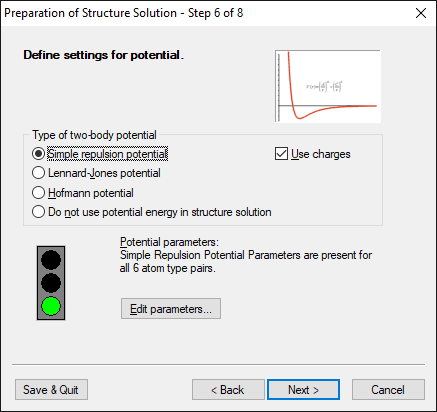 |
Step 6 of 8:
Define settings for potential
This page is dedicated to the input of data needed for the calculation of the
potential energy. There are three possible functions that may be used, two
of them - the 'simple repulsion potential' and the 'Lennard-Jones potential' -
need empirical parameters. The "Hofmann potential", introduced in version 1.3,
enables much easier solution of crystal structures of rigid molecules and is
even capable of predicting the crystal structures of rigid molecules without
using any powder diffraction data. So it does not contain any parameters that have
to be adjusted by the user, making its usage extremely
simple. Concerning the other two potentials, Endeavour has been designed
carefully to especially make the generation of potential parameters as simple
as possible.
|
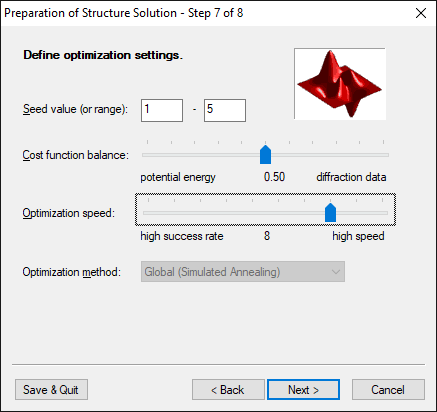 |
Step 7 of 8:
Define optimization settings
This page offers some options concerning the optimization. For instance, the
so-called 'cost function balance' may be adjusted, i.e. the contribution of
either of the two parts of the cost function to the overall value. Thus, weak
diffraction data may to a certain degree be balanced by a high quality
potential and vice versa.
Besides this, the speed of the calculation is controlled by the slider
'optimization speed' that could take any integer value from 1 to 10. A value of
10 means highest optimization speed, however, because of the underlying
simulated annealing optimization method, also lowest success probability.
|
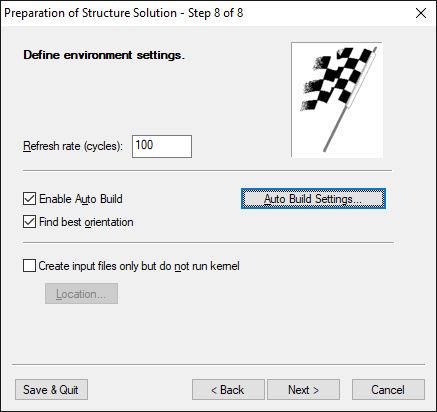 |
Step 8 of 8:
Define environment settings
Since Endeavour is not only a program for structure solution but also for
structure visualization, you can view the intermediate structures during the
structure solution process. On wizard page 7 you can tell the program how this
should be done. |
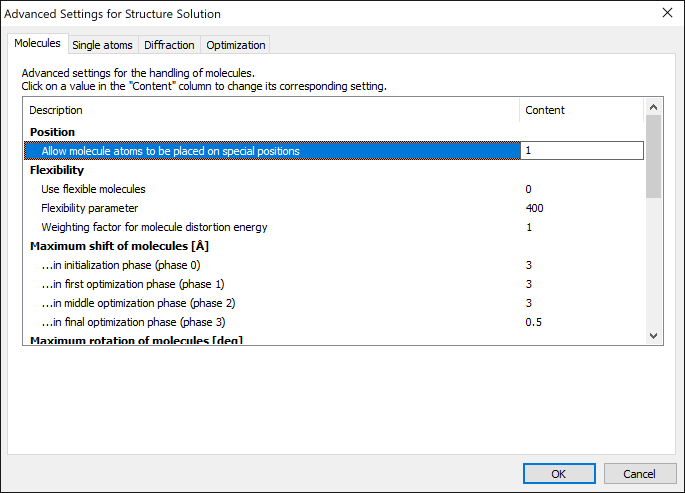
Since version 1.5, you can check out several more parameters through the "Advanced settings for structure solution" dialog,
which is available from the final page of the structure solution wizard.
For example, you can treat molecules as flexible rather than rigid-body molecules or - which is new in version 1.8 - allow
the atoms of a molecule to be placed on special positions:
Endeavour Features Overview...
Next: Space-group Determination...
|