
|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Building up structural partsAdding Some or All Atoms (and Bonds) DirectlyWhile the generation of atoms within a well-defined range of the crystal structure lattice is a quite suitable way to get an overview, atoms can be generated directly by applying the corresponding atom code. In most cases the purpose of generating special atoms by code is to create one or more atoms which serve as starting atom of a more complicated inorganic framework or as starting point for molecules. This kind of intialization of the structure picture is mostly followed by functions which complete coordination spheres subsequently or complete fragments. (See the articles "Filling spheres" and "Generating molecules".) In Diamond, you can specify the atoms to be generated by codes or code ranges or - more sophisticated - by selecting atoms from the parameter list and - as far as necessary - symmetry matrices, Bravais translations, and integer cell translations. This is done with the Add Atoms dialog, which is described in details in "Adding Some Atoms from Parameter List". If the atomic parameter list describes one or more complete molecules, e.g. a protein which has been imported from Protein Data Bank, the Add all atoms command can be used to transform all atoms from the parameter list immediately without applying any symmetry operations. See "Adding All Atoms of Parameter List".
Previous article: Support for disorder parts Adding Some Atoms from Parameter List
Diamond offers the comprehensive Add Atoms dialog, where you can specify the atoms to be generated by codes or code ranges or - more sophisticated - by selecting atoms from the parameter list and - as far as necessary - symmetry matrices, Bravais translations, and integer cell translations. To open the Add Atoms dialog, choose the Add Atoms command from the Build menu.
Alternatively you can use the (red marked) button
There are two different ways to define the codes of the atoms you want to be generated:
1. Define the codes directly using the ORTEP-like atom codes.
If you are familiar with this kind of coding atoms, you can enter the code(s) in the input field Code(s),
and then start the generation with OK or Apply Now. 2. Define the codes indirectly using the list of atomic parameters, symmetry operations, Bravais translations and the input fields for the ranges of integer cell translations, and then start the generation with OK or Apply Now.
(You may combine these two methods, that means you may select atoms from the lists and additionally specify codes.)
Selecting atoms and symmetry operations
All lists in the Add Atoms dialog work with extended selection. To generate one or more atoms, do the following steps:
1. In the list Atom site list, select the atom(s) you want to be generated.
2. In the lists Symmetry operations and Bravais translations, select the symmetry matrices and Bravais translations, rsp. If no symmetry operation is to be applied, select "x,y,z" and "+(0,0,0)", rsp. (which is the default setting in these lists). If needed, multiple symmetry operations and/or multiple Bravais translations can be applied to the selected atom(s).
3. In the six input fields below, enter the integer cell translations, as far as needed. If you need not apply cell translations, leave these fields untouched. (The default values when opening the dialog are 0 for all cell translations.) 4. Start the generation of the atoms by closing the dialog with OK. If you want to generate more atoms subsequently, you should use the Apply Now button instead, since this does not close the dialog. (Use the Reset selections button, if you want to reset the settings.)
Defining atom codes
1. Enter the atom codes into the input field Code(s). Atom codes may be defined as single, multiple codes (separated by commas, semicolons or blanks) or as code ranges.
Example: "3454011-4556042" creates atoms with numbers #3 through #4 (positions in the parameter list; first position is #1) and applies the symmetry operations #1 through #4 and Bravais translations #1 through #2 as well as the integer cell translations -1 <= x <= 0 and -1 <= z <= 1 to these atoms. This code range will generate 2 * 4 * 2 * 2 * 3 = 96 atoms. 2. Start the generation of the atoms by closing the dialog with OK. If you want to generate more atoms subsequently, you should use the Apply Now button instead, since this does not close the dialog.
Filter will not be used
When Diamond generates atoms from the Add Atoms dialog, the settings in the filter (see "Filter") will not be taken into account.
Multiple atoms on special sites When generating atoms via the Add Atoms dialog, Diamond does not check, if multiple atoms are generated on one and the same position. This may happen, if you apply multiple symmetry operations to one or more atoms on special sites.
Adding All Atoms (and Bonds) of the Parameter List(s)Crystal structure descriptions of molecular structures often contain the coordinates of those atoms, that together just describe a molecular unit. To get a complete molecule only these atom positions are needed, that means the application of symmetry operations is not necessary. This strategy can save much time for calculation, especially with very large structures (e.g. proteins). To add all atoms from the parameter list (and optionally bonds, H-bonds and contacts, if corresponding connection parameters are given), choose the Add All Atoms and Connections command from the Build menu.
Alternatively you can use the (red marked) button Unlike the Add All Atoms and Connections dialog that is opened by the command from the Build menu, the toolbar/accelerator command creates all atoms from the atomic parameter list as well as all bonds from the bond parameter list but neither H-bonds nor contacts from their corresponding connection parameter lists, whereas the dialog optionally considers also H-bond and non-bonding contact parameters. When you apply the Add All Atoms (...) function only to the atomic parameter list containing N atom sites (or when there are no connection parameters available), this procedure creates N atoms directly without applying symmetry operations, Bravais translations, and integer cell translations. That means, all N atoms have the code A555011.
Like for the generation of atoms from the "Add Atoms and Connections..." command/dialog, the settings of the filter (see "Filter") will not be taken into account. Add Atoms Directly from the atomic parameter tableThe Add Atoms Directly command is available from the context menu of the atomic parameters table and adds the atoms that are currently marked as selected in the atomic parameter table directly - that means without involving additional symmetry operations or cell translations - to the current structure picture. With other words, all (marked) atoms of the parameter list are created at their "x, y, z" positions. This command is suitable, if the parameter list (or part of it) describes one or more complete molecules.
Previous article: Support for disorder parts [1] COD: 1004001. Jin, Song.; Zhou, Ran.; Scheuer, Ellen. M.; Adamchuk, Jennifer.; Rayburn, Lori. L.; DiSalvo, Francis. J.; "Synthesis, Characterization, and Ligand Exchange Studies of W6S8L6 Cluster Compounds"; Inorganic Chemistry, 40, 2666-2674 (2001) |
|
Page last modified July 9, 2022. Copyright © 2022 Crystal Impact GbR. All rights reserved. Contact Webmaster |
 in the Build toolbar.
in the Build toolbar.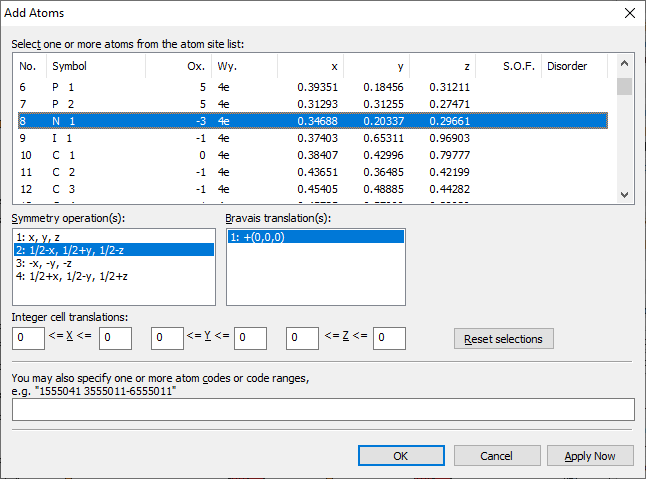
 button from the Build toolbar
or use the accelerator key Shift+Ctrl+A.
button from the Build toolbar
or use the accelerator key Shift+Ctrl+A.