|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Building up structural partsGenerating Molecules
Previous article: Creating and modifying broken-off bonds Overview
This article and the following articles describe how molecules can be created from the parameter list or how molecular fragments can be completed.
This first article shows you how the molecular generator of Diamond works and how to process infinite structures or macromolecules: You can define/change molecular units from the atomic parameter list basing upon connectivity or atomic environments:
The molecule cage can limit the molecular generator for macromolecules that are "too big" or for infinite structures:
And it describes how molecules can be generated from the atomic parameter list:
The following articles will tell you how to complete molecular fragments (for example
after a "Fill unit cell" command), and how to create packing diagrams: The table of molecules is described in the article "Tables of objects like atoms, bonds, molecules etc."
How the Molecular Generator Works
The creation of molecules is comparable with the cyclic filling of coordination spheres. For molecules the number of cycles is in principle unlimited, and the procession will stop after the last atom of each molecule has been created.
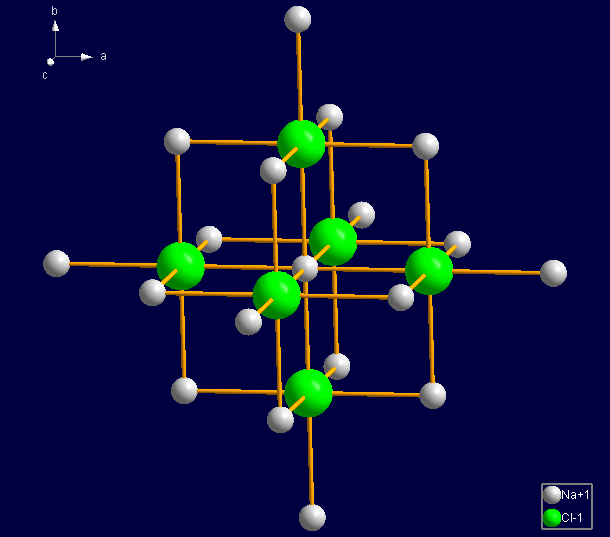
That means if you have for example created the contents of the unit cell, and then apply the molecular generator, Diamond completes the coordination spheres for every atom, and in every further cycle it completes the coordination spheres of the atoms that have been created in the cycle before. If the structure has infinite parts, what is the case in most of the inorganic structures, the molecular generator would not come to an end. To avoid this, the molecular generator stops when it encounters the same atom/bond patterns in neighbouring cells. Besides this, Diamond offers several settings, the molecule cage, to bring the generation of molecules to an end. These settings are described in the following article "Molecule Cage: Special Settings for the Molecular Generator". If you apply the molecular generation to typical inorganic frameworks, for example to sodium chloride, the results are unpredictable.
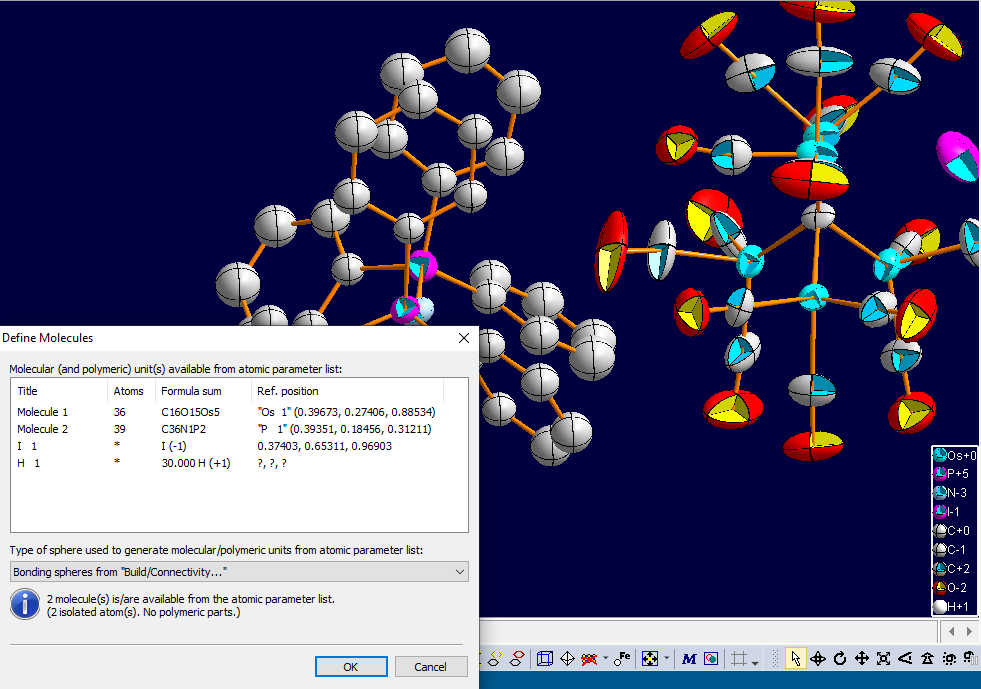
You can stop the molecular generator with the Ctrl+Break key combination. Please note that a proper setting of the connectivity is an essential requirement for the molecular generator. Otherwise a structure may become infinite, although it has isolated molecules, if "wrong" bonding spheres have been defined. To check the connectivity, choose the Connectivity command from the Build menu. Definition of Molecular UnitsSince version 4, Diamond automatically defines (one or more) molecular units from the atomic parameter list, basing upon connectivity (default) or atomic environments. The command and dialog "Build/Molecules/Define Molecules..." can be used to check (and change) the composition of these molecular units. The following screen shot shows the dialog for the sample file "OSPNI_1_rendered.diamdoc", containing two molecules, one isolated I atom as well as 30 H atoms whose positions are not determined in the data set. The numbers of atoms, the formula sum and the molecule's reference atom each is given in the columns of the table "Molecular (and polymeric units) available from atomic parameter list".
By default, the type of sphere used to generate molecular units, are the bonding spheres from the connectivity, see: Connectivity, part 1: Bonding spheres. You can change this option to "atomic environments", see: "Atomic environment" as additional (optional) criterion when filling a coordination sphere or adding coordination polyhedra. Molecule Cage: Special Settings for the Molecular Generator
Diamond offers settings, the molecule cage, to limit the molecular generator for macromolecules that are "too big" or to infinite structures.
1. The number of recursions can be limited to a maximum number of cycles during the recursive filling of coordination spheres.
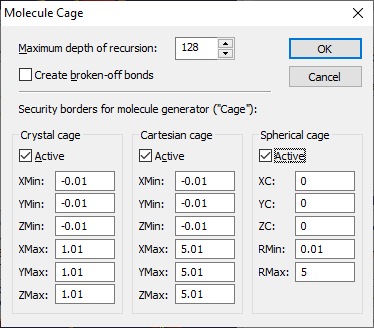
2. Security borders, which can have the shape of spheres, boxes or parallel epipeds, can be defined to stop the generation of molecules at well-defined borders. These security borders are called "cage" in Diamond. To edit the settings for the molecular generator, choose the command Build/Molecules/Get Molecules..., which opens the Get Molecules dialog. In the dialog, click on the Cage... button to open the Molecule Cage dialog:
The input field Maximum depth of recursion defines the number of cycles Diamond uses to create the atoms in the coordination spheres of those atoms that have been created in the previous cycle. If this number is reached during a molecular generation process, the structure may be polymeric. The maximum value is 128. If the number is too small or the molecule is too big or the structure is really infinite, you can call the molecular generator again starting with the previously created molecular fragment. For more details how to complete molecular fragments, see the article "Completing Molecular Fragments".
The other settings define the security borders. Diamond uses three kinds of security borders ("cages"):
Boxes and sphere may be activated independent from each other. The molecular generator will stop searching for neighbours, if an atom found lies outside at least one of these bodies. Atoms outside the boxes and/or sphere will be created but do not serve as starting point for a search for neighbours. Generating Molecules from the Atomic Parameter List
Diamond offers two strategies to create one or more molecules from the atoms in the parameter list:
(1) By adding all atoms of the parameter list without applying any symmetry operations. This is the method of choice if the atomic parameter list describes one or more entire molecules.
(2) By creating molecules from every atom of the parameter list applying symmetry operations automatically if necessary. This is the question of choice, if the parameter list does not describe complete molecules, for example if the molecules have internal symmetry.
To create molecules directly from the atomic parameter list (method #1), do the following steps:
1. From the Build menu, choose the Add All Atoms (and Connections) command, or use the "hotkey" Shift+Ctrl+A,
or press the (red marked) button 2. Check the connectivity settings for the creation of bonds which follow in the last step. Compare the article "Connectivity" how to check or change the connectivity.
3. From the Build menu, choose the Connect Atoms command, or use the "hotkey" Shift+Ctrl+N,
or press the
If you do not know if the atomic parameter list describe complete molecules or if you know definitely that molecules have internal symmetry elements and the atomic parameter list contains the asymmetric parts only ("asymmetric unit"), you must use method #2. To create molecules from the parameter list, do the following steps: 1. Check the connectivity settings.
2. From the Build, choose the Get Molecules command from the Molecules sub-menu, or use the "hotkey" Shift+Ctrl+M,
or press the
Method #2 is the secure method, since it applies symmetry operations to create the symmetry equivalent parts of the molecules from the asymmetric unit. When Diamond generates molecules from the parameter list, it uses the following procedure: 1. For the first atom in the parameter list, create an atom without applying any symmetry operation. That means the first created atom will have the atom code "1555011".
2. With this atom as starting point, call the molecular generator. This will recursively fill the coordination spheres around the starting atom.
3. Repeat these steps for the next atom of the parameter list, but only if that atom or a symmetry equivalent atom has not yet been created.
If the atomic parameter list describes one complete molecule, step #3 will create no further atom, since all atoms of the molecule have been created in step #2. If the atomic parameter list contains more than one molecule, step #3 will be called only as often as molecules are in the parameter list.
The following example shows a molecule of cytochrome C (from rice embryos (ORYZA SATIVA L); Protein Data Bank entry code 1CCR), which has been created using method #1. Method #2 would lead to the same result:
Another example uses the gamma modification of cyclo-octasulfur from the Inorganic Crystal Structure Database (Collection Code: 66517). The structural parameters from the data sheet are:
Bibliographic data
Name
Cyclo-octasulfur - g
Formula
S8
Author(s)
Gallacher, A C, Pinkerton, A A
Title
A redetermination of monoclinic g-Sulfur
Journal
ACSCEE 49 (1993), pp. 125 - 126
Crystal data
Formula sum S8
Crystal system monoclinic
Space group P 1 2/c 1 (no. 13)
Unit cell dimensions a = 8.4375(30) Å
b = 13.000(2) Å
c = 9.250(3) Å
b = 124.50(3) °
Cell volume 838.80(1725) Å3
Z 4
RAll 0.036
Pearson code mP32
Formula type N
Wyckoff sequence g8
Atomic coordinates
Atom Ox. Wyck. x y z
S1 +0 4g 0.64844(20) 0.34375(10) 0.32422(20)
S2 +0 4g 0.80859(10) 0.57813(10) 0.4707(1)
S3 +0 4g 0.74219(20) 0.44141(10) 0.53125(10)
S4 +0 4g 0.58203(20) 0.67578(10) 0.38281(10)
S5 +0 4g 0.07959(20) 0.79688(10) 0.19922(20)
S6 +0 4g 0.24219(20) 0.03088(10) 0.21973(20)
S7 +0 4g 0.30664(20) 0.89063(10) 0.34961(20)
S8 +0 4g 0.14746(20) 0.12695(10) 0.32813(20)
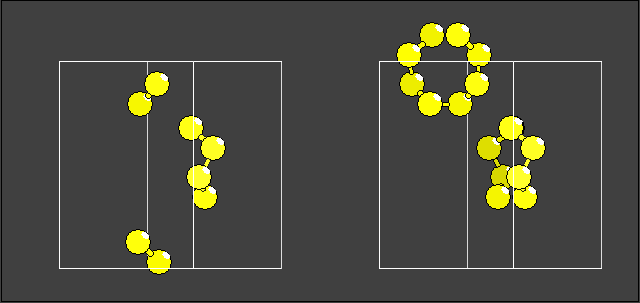
The atomic parameter list contains eight sulfur atoms, but using method #1 leads to the result (left part of the picture below), whereas method #2 leads to the right picture, which shows two molecules with eight atoms each. The atoms in the parameter list above describe only the asymmetric parts of the two molecules:
The list of created atoms for the right picture contains the following atoms:
4 S1 1555011 x, y, z 0.64844 0.34375 0.32422
5 S1 1655021 1-x, y, 0.5-z 0.35156 0.34375 0.17578
6 S3 3555011 x, y, z 0.74219 0.44141 0.53125
7 S3 3655021 1-x, y, 0.5-z 0.25781 0.44141-0.03125
8 S2 2555011 x, y, z 0.80859 0.57813 0.47070
9 S2 2655021 1-x, y, 0.5-z 0.19141 0.57813 0.02930
10 S4 4555011 x, y, z 0.58203 0.67578 0.38281
11 S4 4655021 1-x, y, 0.5-z 0.41797 0.67578 0.11719
12 S5 5555011 x, y, z 0.07959 0.79688 0.19922
13 S5 5555021 -x, y, 0.5-z -0.07959 0.79688 0.30078
14 S7 7555011 x, y, z 0.30664 0.89063 0.34961
15 S7 7555021 -x, y, 0.5-z -0.30664 0.89063 0.15039
16 S6 6565011 x, 1+y, z 0.24219 1.03088 0.21973
17 S6 6565021 -x, 1+y, 0.5-z -0.24219 1.03088 0.28027
18 S8 8565011 x, 1+y, z 0.14746 1.12695 0.32813
19 S8 8565021 -x, 1+y, 0.5-z -0.14746 1.12695 0.17188
Previous article: Creating and modifying broken-off bonds |
|
Page last modified August 10, 2022. Copyright © 2022 Crystal Impact GbR. All rights reserved. Contact Webmaster |
 in the Build toolbar.
in the Build toolbar. button in the Build toolbar.
button in the Build toolbar. button in the Build toolbar.
button in the Build toolbar.