|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Editing structural dataEntering a new structure
Read in this article:
Previous article: Saving and exporting structure data Creating a new documentThis part of this article will describe how to create a new document and if and how far to create a new structure for this new document.
The command of choice to create a new document is the New... command in the File main menu. The New Document dialog prompts you The alternative is to create a new document through the toolbar or with the accelerator key Ctrl+N. This accelerator command directly creates a blank document, starting with an empty structure table (i.e. there are no structure data at all in that new document), and the above mentioned dialog is not involved.
If you start with a blank document, the first view will be an empty structure table. You can later use the New Structure command from the Structure menu to add the first structure data set and type in structure parameters manually, or you can use the Insert From File or the From Structure Type Database command from the Structure menu to insert one or more structure parameter sets or a data set from the structure type database, rsp., into the new blank document. Creating a new structureThis part of this article will describe how to create a new structure for a new document, i.e. the first structure for a new document, and if and how far to enter basic data, such as cell, space group and atomic parameters for this new structure. If you create the new document with the above described New command from the File main menu, you can choose between three options in the New Document dialog: Typing in structure parameters Inserting data set(s) from a file
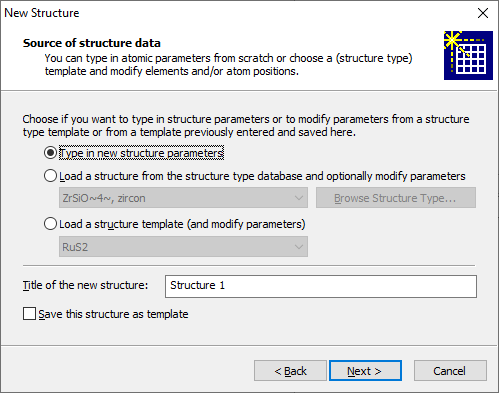
Loading a structure from the structure type database If you decided to start with a blank new document, you can use the command New Structure from the Structure main menu to add the first structure data set to that document. This command is also the choice to add another structure data set to a Diamond document that already contains one or more data sets. This is described below. Using the New Structure Assistant to enter structure parametersThe New Structure Assistant guides you through the input of the required data for a new crystal structure (space group, unit cell, atomic parameters) or molecular structure (atomic parameters only) and gives you some options, if and how to start a first picture for this new structure data set. You can enter these structure parameters from scratch or access pre-defined structure data from the structure type database or from a structure template and modify these parameters, e.g. change atom types and/or positions. The New Structure Assistant is called through the Structure -> New Structure command or in context with the above mentioned New Document dialog, called from the File -> New command. The first page ("Welcome page") gives you a summary what you can expect during the next steps. Proceeding to the next page using the Next button shows you the Source of structure data page of the assistant where you choose, if you type in structure parameters and add atom by atom or choose and modify data from a pre-defined structure data set.
To type in structure parameters activate the corresponding radio button. In this case Diamond starts with a blank structure data set. You will define cell parameters and space group on the following page (or none, if you prefer a non-crystal structure) and add atomic parameters on the subsequent page. If you do not want to start from scratch but use the structure parameters of a structure data set from the structure type database, choose the corresponding radio button Load a structure from the structure type database [...]. You will have the opportunity to modify cell parameters, space group, and atom types and/or atomic positions on the two subsequent pages. The title of the structure type to be taken is given in the combobox below the radio button. Use the Browse Structure Type button to browse for another structure type's data set or if no data set has been selected up to now (e.g. if you run this dialog for the first time). Alternatively, you can take the structure data from a template that has been stored by the New Structure Assistant in a previous session. You have the chance to modify these data on the next two pages. The default title for the new structure is "Structure 1", you can change the title in the input field Title of the new structure. You can store the structure parameters that will be defined or modified on the subsequent two pages in the Diamond section of the Windows Registry. (The template will be stored with the title given on this page.) So you can access these data in a later call to this assistant under the Load a structure template option. The Next button brings you to the Cell parameters and space group page where you choose to enter a crystal structure with cell parameters and space group or what we simply call a "molecular structure" (just atomic parameters in orthogonal coordinates). If you used the structure type or template option on the previous page, the type of structure and the cell parameters and space group are copied from the structure type or template, rsp.
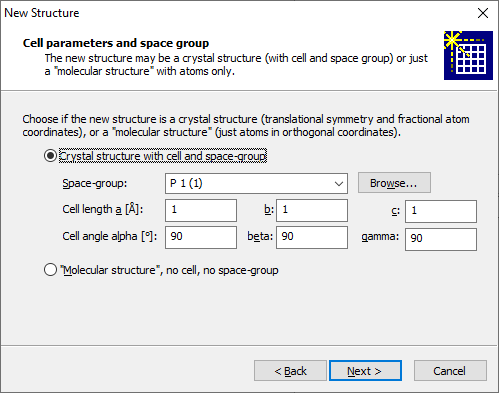
For a crystal structure use the corresponding radio button and enter the space group and the six cell parameters a, b, c, alpha, beta, and gamma into the input fields below. For the space group you can either enter the Hermann-Mauguin symbol of the desired space group directly, or press the Browse... button and select the space group from a list. Depending on the (crystal system of the) space group, one or more input fields of the six cell parameters may be fixed (and deactivated). If your structure has no translational symmetry, i.e. no cell parameters and no space group., choose the Molecular structure radio button instead. Note: In Diamond, we simply call it "molecular structure" - although this does not necessarily mean that the atoms form one or more molecules. It may be any moiety of atoms. The Next button brings you to the page where you enter or modify the atomic parameter list. If you started from a blank structure, this list is empty. you will have corresponding input fields and a list below with the atoms defined so far:
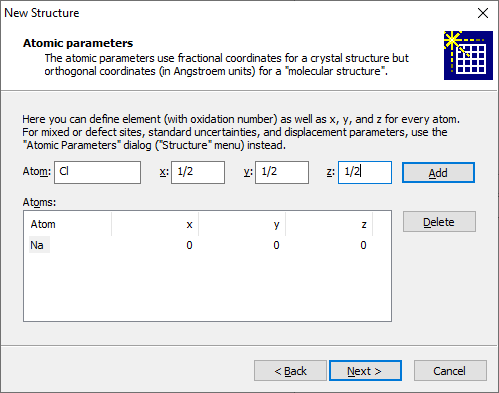
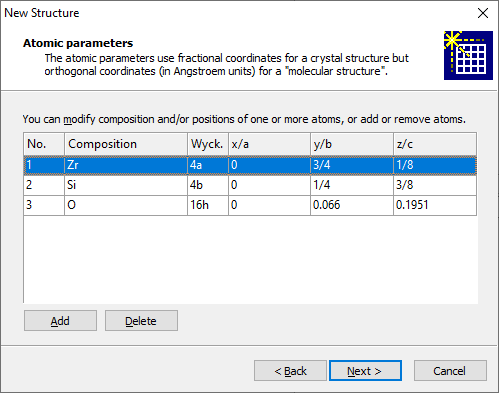
In the screenshot above, we have already entered a Na atom at (0, 0, 0) and are about to add a Cl atom at (1/2, 1/2, 1/2). In order to enter the parameters of an atom, please enter the element (e.g. "na") into the input field Atom. If you would like to define a charge/oxidation state for the atom, you have to enter it together with the element (e.g. si+4, o-2). Afterwards, enter the atomic coordinates (x/a, y/b, z/c for a crystal, x, y, z for a molecular structure). Finally, press the Add button to add the atom to the list below. If you would like to delete an atom from the parameter list, please mark the corresponding atom in the list, then press the Delete button. If your source of structure data was loaded from the structure type database or from a template, you will see a report of already (pre-)defined atoms whose composition and/or coordinates you can modify here.
For this screenshot we made use of the "Load a structure from the structure type database" option described above. So the three atoms of the parameter list of ZrSiO4 (zircon) appear here. To modify atoms of a pre-defined structure data set, click on the item you want to change, i.e. the atom's composition or the atom's position. You can choose a different site from the Wyckoff positions' drop-down list. You can define a charge or oxidation state behind the element symbol and a site occupation factor before the element symbol. You may even define a mixed site, e.g. "0.5Na+1 + 0.5K+1". (Use blanks before and after the '+'symbol between the components.) You can use the Add button to add another site to the atomic parameter list or Delete to remove the selected atom(s) from the parameter list. When you are done with entering or modifying the atoms of the atomic parameter list, click on Next to come to the Picture creation page of the assistant. For a crystal structure the following options are offered:
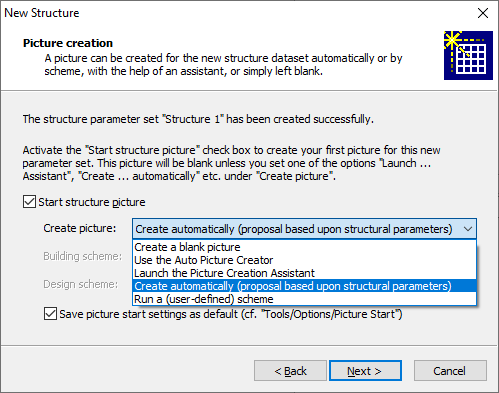
By default, Diamond creates a first structure picture for the structure data set you have entered on the previous pages. Since it is not mandatory to have a structure picture assigned to a structure data set, you can clear the checkmark at Start structure picture. In this case, you can add your first picture later using the command Start Picture from the Picture menu. If you decide to let Diamond create your first structure picture, you have further the choice to leave this picture blank and build it up manually or let Diamond create a picture automatically from a structure proposal or by guidance or by a user-defined scheme. The options are available from the dropdown box at Create picture: Create a blank picture Use the Auto Picture Creator Launch the Picture Creation Assistant Create automatically (proposal based on structural parameters) Run a user-defined scheme
Save picture start settings as default Picture creation for a molecular structure If you have defined a molecular rather than a crystal structure, there is only the choice to start a first picture or not and - if yes - to create all atoms from the atomic parameter list or not. If the Create all atoms checkbox is checked (default: yes), all atoms defined on the previous page will be created in the structure picture. This is similar like running the Add All Atoms (and Connections) command from the Build menu. Note: The Add All Atoms (and Connections) command creates all atoms from the atom parameter list and (default setting) all bonds from the bond parameter list. Since you only enter atoms but no bonds in the New Structure Assistant, no bonds will be created automatically when you close this assistant with Finish. Clicking on the Next button will lead you to the last page ("Completion page") of the New Structure Assistant where you have a summary what Diamond will do and which additional or alternative options you have to post-process the structure data and/or the first structure picture. Clicking on the Finish button on the "Completion page" will launch the creation of the structure data set and (optionally) the first structure picture for it. Adding another structure to a documentSince a Diamond document can store the parameters for more than just one crystal or molecular structure, we will show here how to insert another structure parameter set into your open Diamond document.
Entering a new structure data set
Inserting structure data from a file
Inserting structure data from a structure type database Regardless, how and where you take the structure data from, you can use the commands in the Structure menu to modify the structure parameters, as presented in the following articles. Separating a structureDiamond supports multiple pictures for one structure parameter set each. When you have defined multiple pictures and want to change structural parameters for one of the pictures only, you can use the Structure -> Separate... command to define a separate set of structural parameters with this associated single picture. Changing structure parameters for this separated structure parameter set does not reflect the residual N-1 pictures for the "original" structure. The Separate command detaches the active structure picture from the active structure parameter set, creates a copy of the parameter set and assigns the picture to that copy.
Previous article: Saving and exporting structure data |
|
Page last modified October 04, 2023. Copyright © 2023 Crystal Impact GbR. All rights reserved. Contact Webmaster |